
2026 Medical Device Regulations: EUDAMED, FDA QMSR, and Compliance Risk
It’s time to get your regulatory systems in check in 2026.
With the regulatory changes going into effect this year, it’s becoming easier for regulators to see inconsistencies in your data and easier for inspectors to test whether your quality system actually works.
Two developments drive this shift:
- In the European Union, EUDAMED becomes mandatory under the Medical Device Regulation
- In the United States, the FDA’s Quality Management System Regulation moves inspections toward a structure aligned with ISO 13485
These regulatory changes will affect product portfolios, inspection exposure, and how companies organize their teams.
Let’s discuss the changes that are coming and the effect they’ll have on your business.
Table of Contents
In Europe, Your Device Data Becomes Easier to Cross-Check
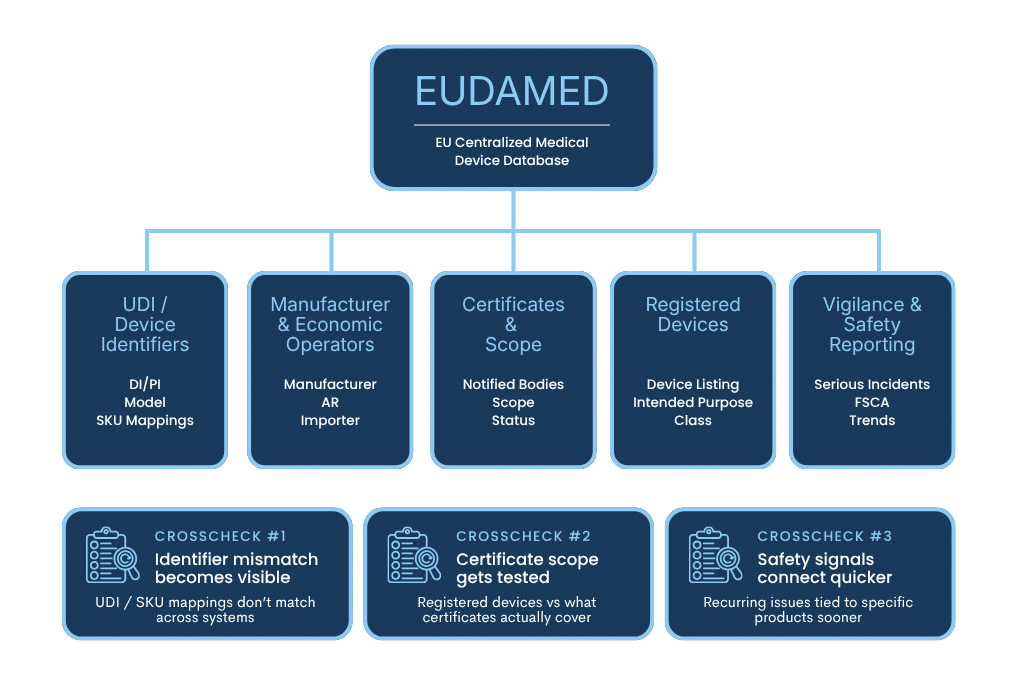
EUDAMED is the EU’s centralized database for medical devices. It holds information about manufacturers, device identifiers, certificates, and safety reporting.
The database itself is not new. What changes in 2026 is that its use becomes mandatory in its active modules.
Why does that matter? Because standardized data makes comparison easier.
Before this shift, information often lived in separate systems. Different formats. Different timing. Different national workflows. If regulators had concerns, they requested records and pieced the story together.
With a centralized system, the starting point is already structured. That reduces the time between a signal and a follow-up question from an inspector.
What This Means for Companies
If your device identifiers do not match across systems, that inconsistency becomes easier to detect.
If your certificate scope does not clearly align with registered devices, that becomes easier to question.
If safety reports show recurring issues, regulators can connect those signals to specific products quicker.
This doesn’t mean automatic penalties, but it does mean there’s less room for messy internal data.
For companies with large or acquired portfolios, that’s where pressure builds. Older products, legacy systems, and relabeled SKUs often carry small inconsistencies that were never urgent. Centralized visibility will make them urgent.
With increased awareness of small inconsistencies comes the risk for more financial consequences.
Compliance Costs Now Shape Which Products Stay on the Market
Under the EU Medical Device Regulation, keeping a device on the market is more demanding than it was under the old MDD framework.
For many products, that means:
- Updating clinical evaluation reports with stronger evidence
- Building more structured post-market surveillance programs
- Preparing periodic safety update reports
- Paying for Notified Body review and certificate renewal under MDR
None of those tasks are unusual on their own. The difference is depth and frequency. MDR expects more documentation, tighter monitoring, and more formal review cycles.
To help companies in the short term, Regulation (EU) 2023/607 extended certain transition deadlines to reduce the risk of device shortages.
That extension gives manufacturers more time, but it does not reduce the amount of work required to maintain compliance in the future.
Why This Becomes a Financial Decision
Every one of those requirements carries cost:
- Internal regulatory and clinical labor
- External consultant support
- Notified Body fees
- Ongoing surveillance and reporting effort
For high-revenue, strategically important devices, that investment makes sense. For low-volume or legacy products, it may not.
Imagine a product that generates modest annual revenue in the EU but now requires updated clinical evidence, new technical documentation formatting, and ongoing MDR surveillance commitments. When leadership calculates total remediation and maintenance cost over several years, the return may no longer justify the effort.
That is where portfolio rationalization begins.
The Shift That Matters
Under the previous system, regulatory upkeep was often treated as overhead. A fixed cost of doing business. Under MDR, regulatory effort varies significantly by product. Higher-risk classifications, older evidence bases, and weaker documentation histories demand more work.
That forces a sharper question:
Is this product strategically important enough to justify the compliance burden?
In many companies, that question is now asked on a product by product basis, not portfolio wide. The result of these regulation changes isn’t dramatic public announcements, but quiet decisions to withdraw certain SKUs from specific markets or to avoid recertifying aging product lines. The regulation itself doesn’t force those exits. The economics do.
In The U.S., Inspections Become More About System Logic
This year, the FDA’s Quality Management System Regulation aligns the U.S. framework with ISO 13485. Here’s what that means in simple terms: Inspectors are less focused on whether a procedure exists. They are more focused on whether your system behaves consistently from start to finish.
Imagine this scenario:
Complaints show that a device component is failing earlier than expected.
An investigator will likely ask:
- Did your risk assessment anticipate this failure?
- If not, why not?
- How were suppliers monitored?
- What corrective action did you take?
- Did that action actually reduce the failure rate?
Each of those questions is reasonable. The pressure comes from the connection between them.
If your complaint files say one thing, your risk file says another, and your CAPA documentation says the issue is closed while complaints continue, that looks like a problem with your system.
Under the ISO-aligned structure, inspectors are more likely to follow that thread all the way through.
For decentralized organizations, this exposes inconsistencies between sites. Different escalation thresholds. Different documentation habits. Different interpretations of risk. These inconsistencies become harder to defend.
Harmonization Reduces Regional “Workarounds”
Regulators globally are moving toward similar expectations for software validation, cybersecurity controls, and post-market monitoring. Companies have historically relied on regional interpretation differences. Something acceptable in one market might receive more scrutiny in another. As expectations converge, weak documentation in one jurisdiction becomes a liability everywhere.
The Real Risk Is Organizational, Not Regulatory
Most compliance failures are not caused by misunderstanding the law. They are caused by unclear ownership.
EUDAMED touches regulatory affairs, labeling, master data, and IT.
QMSR ties together quality, engineering, supplier management, and complaint handling.
If those groups operate in silos, small issues compound:
- A labeling change is not reflected in regulatory records.
- A supplier issue is addressed locally but not fed back into risk documentation.
- A CAPA closes without verifying long-term effectiveness.
These are not dramatic failures. They are everyday coordination gaps. Centralized databases and connected inspection logic simply make those gaps easier to spot.

What Companies Should Be Doing Now
Moving forward, companies will have to be disciplined in their processes to reduce risk exposure. Here are a few ways you can begin to prepare your company for a more disciplined process:
- Reconcile device identifiers across systems before centralized databases expose inconsistencies.
- Test a few real complaint cases end-to-end. Follow the thread from signal to risk file to supplier controls to corrective action. See if the story holds together.
- Attach real compliance cost estimates to product lines rather than assuming all products deserve equal investment.
The European Commission’s guidance on post-market surveillance under MDR provides a practical benchmark for structured monitoring.
The Bottom Line
The 2026 regulatory events do not introduce entirely new obligations. They reduce the tolerance for weak systems.
In Europe, centralized data makes inconsistencies easier to find.
In the U.S., inspections are more likely to test whether your processes connect logically.
Globally, expectations are becoming more consistent.
For medical device companies, the impact shows up in day-to-day operations and in the numbers.
Product portfolios stop being “keep everything unless we’re forced to change.” They become a set of choices. Some devices won’t be worth the ongoing effort to maintain the evidence, paperwork, monitoring, and certification needed to keep them on the market. That pushes clearer decisions about what stays in the EU, what gets updated, and what gets retired.
Inspections shift the same way. It’s not enough for each team to have their own files in order. Regulators will look for alignment across the whole chain. If complaints point to a problem, the risk documentation should reflect it. If a supplier issue is involved, supplier controls should show how it was managed. If a CAPA was opened, the company should be able to show the problem actually improved. When those pieces don’t match, it looks like a company that produces documents but doesn’t control the process.
This is why consistency and follow-through matter so much.
Consistency means the same identifiers, the same definitions, and the same decision rules across teams and sites. Follow-through means records get updated when something changes, not when an audit is coming. It also means ownership is clear: one person or function is accountable for keeping data and handoffs clean.
Teams that treat 2026 as an execution challenge will move through it steadily. Teams that treat it as “a regulatory issue” will spend the year explaining mismatches they should have caught themselves.
If you’re looking for new team members who can operate with consistency within this heavily regulated industry, let our Growth Team help you with your search. We work with clients in the medical device industry daily and have the knowledge, experience and resources needed to find your next hire.
Jack Pollard
Client Support Manager | Growth Practice
(804) 404-7311
jackp@richgroupusa.com


